Wilson Disease
Metabolic, Biliary Atresia & Related Diseases
Description
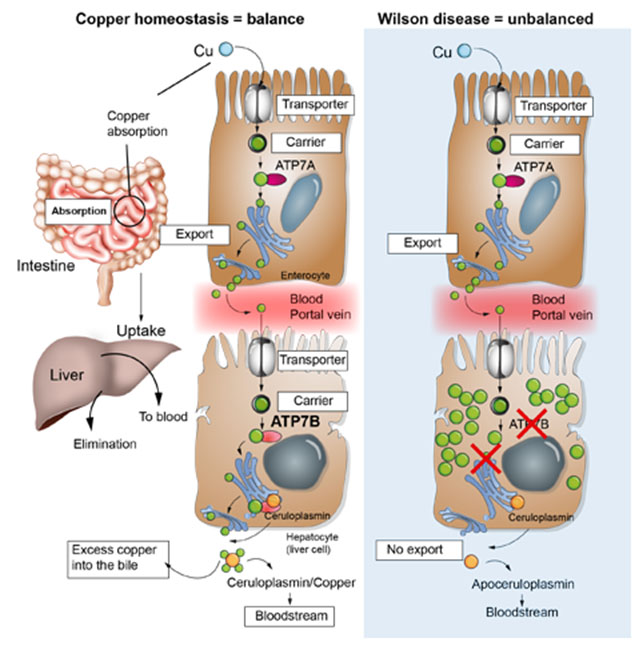
Wilson disease (WD) is a rare autosomal recessive genetic disorder of hepatic copper metabolism. In WD the biliary copper excretion is impaired leading to is accumulation, primary in the liver, and then, when the liver’s capacity for copper storage is exceeded, copper is released into the circulation and may accumulate in other organs, notably the central nervous system, where it may cause neurologic and psychiatric disease. Watch here our educational video on how to manage Wilson disease:

Short educational video on Wilson disease:

Genetics
Wilson disease (WD) is a rare autosomal recessive genetic disorder involving mutations in the ATP7B gene located on the long arm of chromosome 13. The estimated prevalence is of 1 per 30,000 in the general population and the heterozygous status prevalence is estimated in 1/90.
Clinical presentation
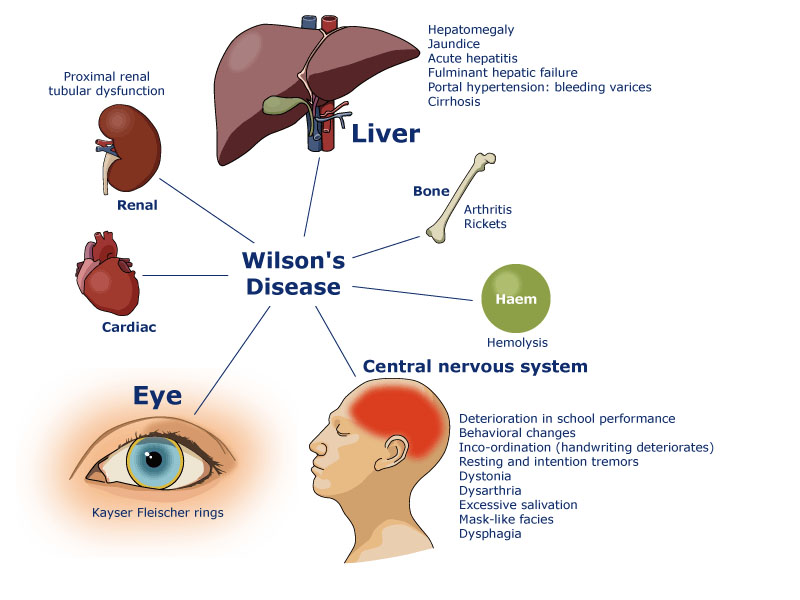
Clinical manifestations are variable, but the key features of Wilson disease are liver disease and neuropsychiatric disturbances. Typically, liver disease is most frequent in children, and as the copper accumulation progress, we can see the first neurological signs appears in the second and third decade of life. Broadly categorized, 40–50% of patients present with some sort of hepatic disorder, 30–40% present with a neurological syndrome, and approximately 20% present with only a psychiatric disorder.
Risk factors
The only known risk factor for Wilson disease is a family history of the disease.
Symptoms
Presenting signs and symptoms of liver disease can be highly variable, ranging from asymptomatic, with only biochemical abnormalities, to overt cirrhosis and fulminant liver failure. Neurological symptoms can be extremely subtle and variable. Most characteristic symptoms are: tremor, dysarthria, dystonia, gait abnormalities and drooling. Psychiatric manifestations may include depression, behavioral changes, mood changes, irritability, impulsivity, declining school performance.
Diagnosis
Diagnosis of WD is based on a pattern of clinical findings, laboratory results and genetic analysis. Serum ceruloplasmin is typically decreased in patients with WD but the predictive value of ceruloplasmin for diagnosis of WD in patients in general is poor. The presence of Keyser-Fleisher rings, total serum copper, non-ceruloplasmin-bound copper, exchangeable, 24-hour urinary copper excretion, brain MR-imaging and genetic analysis are helpful to diagnose WD while a liver biopsy with measurement of hepatic parenchymal copper concentration is required if the clinical signs and noninvasive tests do not allow a final diagnosis or if there is suspicion of other or additional liver pathology. Since none of the available laboratory tests are perfect and specific for WD and typical clinical symptoms may be absent, the combined diagnostic Leipzig score has been adopted in the European Association for the Study of the Liver (EASL) Clinical Practice Guidelines for Wilson disease.
Management
Treatment is based on the removal of copper excess by chelating agents such as D-penicillamine, trientine, or by blocking the intestinal copper absorption with zinc salts. Dietary copper restriction is advised until remission of symptoms and biochemical abnormalities. Treatment regime may vary according to the phase of the disease (initial, stabilized) and the clinical manifestation (asymptomatic, liver/neurologic symptoms). Adherence to medical therapy is a key concern for all WD patients as non-adherence may lead to rapid and sometimes irreversible deterioration. Lifelong follow-up with individually scheduled visits to monitor treatment failures and tailor treatment is necessary. In severe cases, liver transplantation might be indicated.
- EASL Clinical Practice Guidelines: Wilson’s disease. Ferenci P et al. Journal of Hepatology 2012.
- Wilson's Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. Socha et al. J Pediatr Gastroenterol Nutr. 2018.
- Diagnosis and phenotypic classification of Wilson disease. Ferenci P et al. Liver Int. 2003.
- Wilson Disease: Diagnosis, Treatment, and Follow-up. Schilsky ML. Clin Liver Dis. 2017.
Complications
Chronic liver disease due to WD can have the same complications as those classically described in this condition, namely variceal bleeding, ascites, hepatic encephalopathy, hepatocellular carcinoma. Regular monitoring is hence essential to address these potential problems. In extreme cases, liver failure might require liver transplantation. Neurologic deterioration, at diagnosis or in case of treatment disruption, can lead to severe handicaps. Untreated WD evolves invariably to liver failure and death.
CPMS
If you wish to discuss a Wilson disease patient with experts from the ERN, you can upload cases to the CPMS.
The CPMS is essential for interaction between healthcare professionals and experts on clinical decision making, and is provided by the European Union to all ERNs. CPMS supports online multidisciplinary meetings (tele-boards) to discuss patients with diagnostic or therapeutic dilemmas in need of expert consultation. CPMS offers the opportunity to upload and share clinical data of patients and pictures such as histological slides or MRI images. Importantly, this is fully in line with European data protection law. It is obligatory to inform patients about CPMS and obtain their informed consent before their data are entered into the system (see CPMS Privacy Statement published by the European Commission on 14 December 2017). The consent form is available in all EU languages in CPMS.
Media
-

Schematic representation® SwissLiver -

Schematic representation®Eurowilson
Clinical practice guidelines
Please click here to view clinical practice guidelines for the diseases covered by ERN RARE-LIVER.